


Spatially resolved single-cell transcriptomics offers insights into gene expression within tissues, but current technologies are limited by their ability to measure only a small number of genes. To address this, algorithms have been developed to predict or impute the expression of additional genes. These methods often use paired single-cell RNA sequencing data, embedding spatial and RNA-seq data together to make predictions. However, most approaches still need to fully utilize the relational information between genes (like co-expression) or cells (like spatial proximity). Incorporating this relational data could improve the accuracy of gene expression predictions and enhance subsequent biological analyses.
Stanford and Harvard University researchers have developed SPRITE (Spatial Propagation and Reinforcement of Imputed Transcript Expression), a meta-algorithm designed to enhance spatial gene expression predictions. SPRITE refines predictions from existing methods by propagating information across gene correlation networks and spatial neighborhood graphs. This two-step process improves the accuracy of spatial gene expression predictions, leading to better performance in downstream analyses like cell clustering, visualization, and classification. SPRITE can be integrated into spatial transcriptomics data analysis to enhance the quality of inferences based on predicted gene expression.
The SPRITE algorithm was tested using eleven benchmark datasets that combined spatial transcriptomics with RNA-seq data from four species: human, mouse, fruit fly, and axolotl. These datasets, which utilized various technologies and tissue types, were chosen to maintain consistency within species and tissue categories. Before being used, the RNA-seq data underwent normalization and log transformation. SPRITE was evaluated with three spatial gene expression prediction methods—SpaGE, Tangram, and Harmony-kNN—each utilizing distinct approaches for aligning and predicting gene expression. The accuracy of these predictions, both before and following the application of SPRITE, was measured using the PCC and mean absolute error (MAE).
SPRITE operates in two key steps: the “Reinforce” step and the “Smooth” step. The Reinforce step propagates prediction errors across a gene correlation network to correct target gene predictions using an iterative smoothing process. This network is constructed based on Spearman rank correlations between predicted gene expressions. The Smooth step further refines the predictions by propagating them across a spatial neighborhood graph based on the Euclidean distances between cell centroids and adjusted for cell-type similarity. The SPRITE predictions, enhanced through these steps, were evaluated for their impact on downstream analyses such as cell clustering, visualization, and classification, demonstrating improvements in prediction accuracy and the quality of biological inferences.
SPRITE is a meta-algorithm that enhances spatial gene expression predictions by correcting errors through a gene correlation network (“Reinforce”) and smoothing predictions across a spatial neighborhood graph (“Smooth”). Applied to predictions from methods like SpaGE, Tangram, and Harmony-kNN across various datasets, SPRITE generally improved prediction accuracy, reducing mean absolute error and often increasing correlation with ground truth data. Both components of SPRITE are essential, as their combination yields better results. Moreover, SPRITE enhances downstream tasks such as cell clustering, data visualization, and cell type classification, often outperforming models trained on the original measured data.
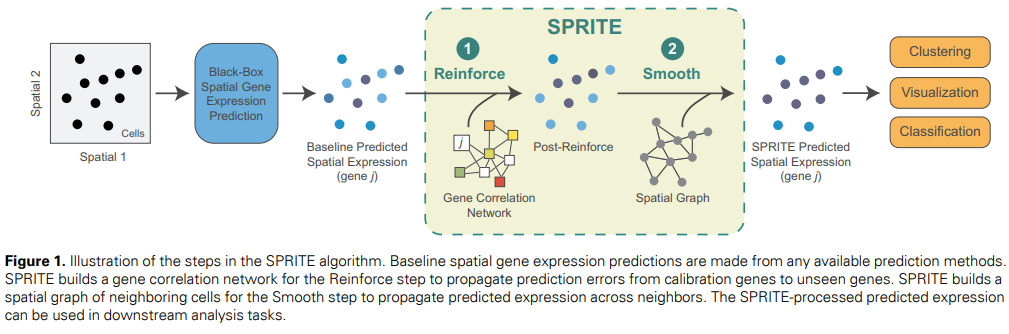
SPRITE is a versatile meta-algorithm designed to enhance spatial gene expression predictions, improving the accuracy of various prediction methods by combining “Reinforce” and “Smooth” steps. It improves gene expression predictions and enhances downstream analyses like cell clustering, visualization, and classification. Surprisingly, SPRITE sometimes outperforms even ground truth data, suggesting it may de-noise gene expression. SPRITE is scalable, with its complexity adjustable by the number of cross-validation folds used. Future research could explore integrating spatial and gene correlation information directly into prediction methods and extending SPRITE to other data types like spatial proteomics.
Check out the Paper and GitHub. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter..
Don’t Forget to join our 47k+ ML SubReddit
Find Upcoming AI Webinars here
The post SPRITE (Spatial Propagation and Reinforcement of Imputed Transcript Expression): Enhancing Spatial Gene Expression Predictions and Downstream Analyses Through Meta-Algorithmic Integration appeared first on MarkTechPost.
#AIPaperSummary #AIShorts #Applications #ArtificialIntelligence #EditorsPick #MachineLearning #Staff #TechNews #Technology [Source: AI Techpark]